BrownDye
Contents
The BrownDye software package consists of 2 simulation programs
and about 25 auxiliary programs for processing data.
For now, it computes the 2nd-order rate constant of the encounter
of two rigid bodies composed of spheres moving according to
Brownian dynamics. It has functionality very similar to
the packages
SDA
and MacroDox.
It includes the following features:
- Bodies composed of charged spheres organized as "atoms" in "residues".
- Uses nested electrostatic grids from
APBS
for computing electrostatic forces.
- Uses hard-sphere interactions for close range (more general close-range
forces to be added soon).
- Uses recursively nested spheres to quickly detect collisions
- Approximate hydrodynamic interactions (sphere-sphere)
- Approximates desolvation forces by Born integrals precomputed on grids.
- When appropriate, lumps charges together to save time.
- Uses an adaptive time step size.
- Uses an new version of the Northrup-Allison-McCammon algorithm that
does not require specification of an outer q-radius.
- Reaction criteria defined by atom-atom distances between molecules.
- Rates can be computed by running one trajectory at a time, or by
using Weighted-Ensemble Dynamics.
- Confidence intervals are given for the rates.
- Can run on multiple processors sharing memory, using pthreads.
- Can output trajectory files for viewing with VTK
- Can compute the probability of the molecules moving from one binding mode
to another
- Automatically generates the b-radius for use with the enhanced
Northrup-Allison-McCammon algorithm.
- Allows a choice between hard-sphere and soft-sphere
repulsive (r - 12) forces for
short-ranged interactions
Browndye
runs under Linux-based operating systems; I haven't tested
it on other Unix systems. It is assumed that you have a C++
compiler, and the makefiles use the GNU C++ compiler g++ (default)
or clang++. (In Makefile in the main directory,
just change the first line as directed in order to switch compilers).
The compiler for the computer
language Objective Caml (or
Ocaml .
for short) is also required;
it is free and easy to install on most systems.
Finally, pthreads is required for the multithreading; and it
is required even if you are not planning on using more than
one thread (this can be changed if there is sufficient demand).
The various programs in BrownDye communicate with each other
by writing out and reading in files in XML format. The notable
exception is that grids use the
OpenDX
format output by APBS.
This has several advantages. From a user's point of view, the
XML files are mostly self-documenting with descriptive tag
names denoting the purpose of the data. Thus, the files
can be read for information or even changed or replaced with
something generated by the user.
From a programmer's point
of view, one can add and subtract new features and information without
having to rewrite the part of the code that parses the file.
The programs of BrownDye use Expat
and Ocaml Expat internally to read in and parse the XML files; these two
software packages are included in the BrownDye distribution.
For now, the main input file is in XML as well; eventually I want to write a
more user-friendly
Python front-end that will convert equivalent Python code to the input file.
The two simulation programs and two of the auxiliary programs are
written in C++, while the rest of the auxiliary programs are written
in Ocaml .
I chose Ocaml because it is my favorite
language (speed and type safety of C/C++ combined with the conciseness
of Python), and others likely will not have to alter the auxiliary
programs. However, I chose C++ for the actual simulation code
for three reasons. First of all,
there are portions representing fairly complex algorithms that might
be reusable for other software. Also,
other researchers might add more features in
the future, and many more people are familiar with C++ than with Ocaml.
Finally, you can not always count on there being an Ocaml
compiler available on your favorite supercomputer; in that event,
you could always run the auxiliary programs on your own computer,
move files to the supercomputer, and let the C++ code do the heavy
lifting.
Besides the simulation programs and auxiliary programs, there is
a front-end program, bd_top, that reads in the main input
file and runs the auxiliary programs to generate the input files
for the simulation programs. The program bd_top functions
almost like the Unix Make utility in that it will run only
those programs needed to update files that depend on updated files
upstream. So, if you want to change or replace one of the intermediate
XML files with something of your own creation, you can run bd_top
again and only those programs will be run which are necessary for
keeping the other files consistent.
The random number generator is the Mersenne twister; I'm using
code from
Hiroshima University. (Update: BrownDye is now using the code from the C++11 Standard Library).
By default, the same physical units as those used by APBS are
used; namely, Angstroms for length, picoseconds for time, and
kT with T = 298 K as the unit of energy. These defaults can
be changed by using different input options for the various programs.
However, the 2nd-order rate constants are output in units of 1/(M s).
In writing the software, various pieces that might be useful
for others are documented here.
Upon building BrownDye, the executables or links to them
are placed in the bin
directory. You can add that directory to your path, or you can
copy or move the executables elsewhere.
If you have a question about any of the programs, you can type
program -help
and a description will be printed.
The first step is to obtain PQR files of the two molecules. This
can be obtained in various ways; for example, the utility
PDB2PQR is a particularly
useful tool. Then, the PQR file must be converted to an equivalent
XML file using the utility p2r2xml. By convention, the
output XML file is given the extension .pqrxml.
In BrownDye, all molecules are treated as collections of charged
spheres, each of which is numbered sequentially and has an atom type.
Each sphere also
belongs to a "residue", which also are numbered sequentially and
assigned a residue type. This is exactly the information given in
a PQR file. In the following discussions, we will often refer
to the spheres as "atoms", even though the spheres do not necessarily
need to be atoms. Likewise, the "residues" often will mean amino acids
in a protein simulation, but can also represent other groupings.
The next step is to obtain the electrostatic fields for both molecules
in OpenDX-formatted file. The suggested manner for doing this is with
APBS software package, although
UHBD could be used and the output file converted using the included utility
uhbd2dx. Several nested grids may be generated and
used for each molecule. Except for the one outermost grid, each grid
must be completely contained within another grid, and a grid may not
overlap another grid unless it is completely contained within it.
As of 31-July-2012, grids with anisotropic spacing can be used.
Another step is to generate the files used in defining reaction criteria.
The program make_rxn_pairs reads in both XML files generated
from the molecules' PQR files, an generic XML file describing the possible
types of atom pairs for reaction criteria, and a search distance,
and outputs an XML file giving reaction pairs for the specific bimolecular
system. If only one reaction is to be studied, then the program
make_rxn_file can then be used to generate an XML file describing
the reaction pathway for the system. More details can be seen
here .
You then must prepare an input file for the front-end program bd_top;
an example is input.xml.bak in the thrombin example. The
following XML tags are used:
- solvent - information about the solvent (details below)
- protein - when set to
true,
TRUE, or True, uses the program
protein_test_charges to generate test charges for Molecule 1, using the same atoms as those used by SDA. Warning: By default, when
protein_test_charges is set to false or omitted, bd_top calls the
program residue_test_charges to generate the charges, lumping all of
the charges in a residue into one charge. To give each atom a charge, you
must give each atom its own "residue", or use test_charges to generate
a charge from each atom.
- molecule0 - information about Molecule 0 (details below)
- molecule1 - information about Molecule 1 (details below)
- n-threads - number of threads of execution; probably should not be more than the number of processors
- time-step-tolerances - this block contains several dimensionless tolerances that govern the time step size. The
last three parameters are used to set a time step size before any time-step splitting occurs,
while the first tolerance is used to split the time step.
- force - governs the splitting of the adaptive time step in response to changes in force and torque. Default is 0.05.
- collision - Time step is proportional to α L2/D, where α is the tolerance, D is the
mutual diffusivity of the two molecules, and L is the gap between the two molecules as estimated by an ellipsoidal
approximation to their shapes. Default is 0.05.
- reaction - Same as above, except L is the smallest reaction coordinate of the possible reactions leading from the current reaction state. Default is 0.005.
- minimum-dx - average minimum distance moved during a time step. This is proportional to (D Δ t) 1/2, where Δ t is the minimum
time step size. The minimum step size is computed from this. Default is 1.0.
- minimum-reaction-dx - If a reaction coordinate is small, then the minimum step size given by minimum-dx can be overridded by the value given by this tag. In other words, the step step still remains larger than that given by minimum-dx unless the tolerance given by the reaction tag is violated. The default value is that given by minimum-dx. This allows very small time steps near a reactive state.
- hydrodynamic-interactions -
if this is set to
false, False, or FALSE, then no hydrodynamic interactions between the molecules are used. Hydrodynamic interactions are used by default.
- reactions - file containing reaction data
- seed - random number seed
- output - name of output file
- start-at-site -
if this tag exists and contains
true , True , or TRUE ,
then the molecules are
started in relative positions according to the coordinates in the
two pqrxl
files, rather than at the distance given by b-radius.
When this is used, then the the interpretation of the computed 2nd-order
rate constant is not physically meaningful; rather, the probability
of reaction versus escape should be used.
- b-radius - this optionally gives the b-radius of the Northrup-McCammon-Allison algorithm,
which is the starting center-to-center initial separation of the molecules. If this is not given, then a value
is automatically computed as the smallest radius that still gives almost spherically symmetric forces.
- include-desolvation-forces - this optional tag indicates whether the desolvation forces are used.
The default is "true". If the contents are "false", "False", or "FALSE", then the desolvation forces are not used.
- desolvation-parameter - this specifies
a numerical factor by which to multiply the integral computed by the program
born_integral . The default is 1.
The block denoted by the solvent tag has the following tags inside:
- dielectric - dimensionless dielectric (usually 78 for water)
- debye-length - Debye length
- kT - Boltzmann constant times absolute temperature. Default is 1 (remember, units are in 298 K times k).
- water-viscosity - viscosity of water. By default, it is the value in the
default units used by BrownDye. It is useful only if you are changing physical units.
- relative-viscosity - viscosity relative to water. Default is 1
- vacuum-permittivity - permittivity of vacuum. By default, it is the value in the default units used by BrownDye. It is useful only if you are changing physical units.
- use-6-8-force -
if this tag exists and contains
true , True , or TRUE , then the soft atoms interact via a 6-8 force (see below) rather than a 6-12 Lennard-Jones force.
The blocks denoted by tags molecule0 and molecule1
have the following tags inside:
- prefix - prefix used for naming all the intermediate files having to do
with this molecule
- atoms - name of PQRXML file containing atoms
- all-in-surface - if this is true, then all the atoms are included in the surface for collision testing. By default, it is false. (The bug in auxiliary program surface_spheres has been fixed.)
- apbs-grids - one or more APBS electrostatic grids. Within this block
are one or more blocks, denoted by the tag
grid, with the name of the
OpenDX file containing the grid. The grids may be listed in any order.
- solute-dielectric - Dimensionless dielectric constant of solute
- soft-spheres - This is an optional tag pair containing an additional pqrxml file which denotes
spheres that are subject to "soft" short-ranged interactions. In the computation, if two "soft" spheres interact, they do so
using the Lennard-Jones potential:
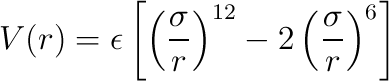 where r is the interatomic distance,
ε is energy well depth, and σ is the sum of the two radii. If at least one of the spheres is not "soft",
then they interact via the hard-sphere interaction. For example, if you want all spheres to be soft, then the file name should
be the same file as contained in the atoms tag. (See auxilliary program
where r is the interatomic distance,
ε is energy well depth, and σ is the sum of the two radii. If at least one of the spheres is not "soft",
then they interact via the hard-sphere interaction. For example, if you want all spheres to be soft, then the file name should
be the same file as contained in the atoms tag. (See auxilliary program make_soft_spheres.)
If the tag use-6-8-force is set to true, then the
force has the form
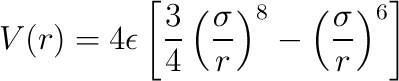
- short-ranged-force-parameters - parameter file for the short-ranged interactions, using the format described
in APBS XML Format section of the APBS user's manual.
This is required if any of the spheres of the molecule are soft. In computing the Lennard-Jones force between two spheres, the value
of ε used is the geometric mean of the value of ε for each atom as given in the file, and the value of σ used is
the sum of the radii as given in the file. Included with BrownDye is the auxiliary program make_repul_params, which can generate
a parameter file for users who just want the repulsive r-12 interaction.
- radii - Because spheres can either be "hard" or "soft", we distinguish between the "hard radius" and "soft radius" of each sphere.
Either radius can come either from the pqrxml file for the molecule, or from the short-ranged parameter file. By default, the
hard radii come from the pqrxml file, and the soft radii come from the parameter file. However, that can be changed. The optional radii
section has the following optional tags:
- hard - gives the source of the hard radii; can have only the values parameter-file or pqrxml-file.
- soft - gives the source of the soft radii in the same manner.
Sometimes it is useful to have "dummy atoms" to denote reaction criteria. If a sphere is given a radius of zero, then it does not interact with other spheres and can freely overlap them. It can still be used to describe reactions (see below ).
- tether -
establishes a tethering spring between an atom on Molecule 0 and one on Molecule 1. This tag pair must appear
inside both the molecule0 tag and the molecule1 tag. Inside the tether
tag pair of Molecule 0, the following
tags must appear:
- atom0 - number of the atom on Molecule 0
- resting-length - length of the spring with no force
- length-deviation - standard deviation of the spring length from its resting length at thermal equilibrium. This is an indirect way
of specifying the spring constant.
Inside the tether tag of Molecule 1, one tag must appear:
- atom1 - number of the atom on Molecule 1
In addition to the tags used in the main block, the following tags are
used in the case of single-trajectory simulations:
- n-trajectories - number of trajectories
- n-trajectories-per-output - number of trajectories between updates of the output file
- max-n-steps - maximum number of time steps taken before giving up and declaring the molecules to be "stuck"
- trajectory-file - prefix of files used for outputting of trajectories.
A different file is created for each thread. If the prefix is "trajectory", then the file for Thread 0 is "trajectory0.xml", and so forth.
This is optional; if it is not given, then no files are generated. The trajectory file consists of
rotation matrices and translation vectors describing the position of Molecule 1 relative to Molecule 0.
- n-steps-per-output - number of time steps per output of system state to the trajectory file. In addition, when the system undergoes a change in reaction state, the first system state of the new reaction state is output, with an additional XML tag new-rxn-state labeling the block of the state.
- min-rxn-dist-file - file to output minimum reaction coordinate for each trajectory (see below).
The following tags are used for weighted-ensemble simulations:
- n-copies - number of system copies
- n-bin-copies - number of system copies used in building bins
- n-steps - total number of steps; if you have m copies and you do n time steps, the total number of steps is n*m
- n-we-steps-per-output - number of time steps between updates of output file
The program bd_top is invoked as such:
bd_top input.xml
This program runs various auxiliary programs and
generates several intermediate files, as described
here .
In addition, it generates a file described as
prefix0-prefix1-simulation.xml, where prefix0
and prefix1 are the file prefices given in the input file.
This is the main input file to the actual simulation programs.
Before running the simulation program,
the user can replace or change any of the
intermediate XML files and rerun bd_top.
In order to run a single-trajectory simulation, the program
nam_simulation is invoked with the input file from bd_top:
nam_simulation prefix0-prefix1-simulation.xml
The main results are periodicly
written to the file specified by the results
tag in the input to bd_top. At any time, the 2nd-order reaction
rate can be computed by running compute_rate_constant:
cat results.xml | compute_rate_constant
assuming that results.xml is the results file.
This outputs the estimate of the rates and their 95% confidence intervals
to standard output.
Trajectories can be viewed by processing the
trajectory files with the auxiliary programs process_trajectories
and xyz_trajectory.
The trajectory files are output in a MIME format within the XML in order to save space, and the completed files include information necessary to quickly select individual trajectories. In addition to the trajectory files themselves, nam_simulation also outputs trajectory index files, whose names follow
the cooresponding trajectory file. For example, if the trajectory file is "traj2.xml", the index file is "traj2.index.xml". The index file is used in order to avoid plowing through megabytes of XML in order to find a particular trajectory.
Each trajectory is given a number (starting at 0), and each trajectory is divided into one or more numbered subtrajectories.
Each time the molecular system completes a reaction, the current subtrajectory terminates, and if the reaction is
not final, a new subtrajectory starts. If there is only
one reaction, then there is only one subtrajectory.
Regardless of the argument of the -nstride flag below, the
last conformation before a reaction is always output; this allows the
user to see the final bound state, for example.
The program process_trajectories takes the following arguments with flags:
-traj - trajectory file from nam_simulation
-index - index file
-n - trajectory number in file (starting with 0)
-sn - subtrajectory number (starting with 0); default is 0.
-nstride - only every nstrideth state is output; the default of 1 means that every state is output
-srxn - find subtrajectories resulting from completion of the given reaction (see below)
-ucomp - find subtrajectories starting from the given state but failing to complete
Output to standard output is an XML file, not in MIME format, of a
single subtrajectory.
In addition, this program lets you select out
subtrajectories that have led to a particular reaction. It is called with
the -traj and -index flags and their file arguments,
and with the flag -srxn with the given reaction name as the argument. This
causes it to print out a list, in XML format, of each trajectory and subtrajectory
that has led to the reaction. In a similar manner, the program will output a list
of subtrajectories starting from a state that lead to an escape; the name of the
state is given as the argument to the -uncomp flag.
Note: This new version of process_trajectories is not backwards-compatible
with older trajectory files. The previous version is included as process_trajectories_old.
The output from process_trajectories can be processed by
xyz_trajectory to generate files that can be viewed. It takes the
following arguments with flags:
-mol0 - molecule0 pqrxml file
-mol1 - molecule1 pqrxml file
-trajf - trajectory file output from process_trajectories
-pqr - output as concatentated PQR files
-trial - outputs only an estimate of the file size in Mbytes.
Output to standard output is a concatenated XYZ file that can be
read and viewed with VMD.
For now, we are stuck with the radii
that VMD assigns to the atom names, so this part is not yet completely
general with respect to arbitrary spheres.
Finally, the -pqr flag will output the trajectories as a collection of concantenated PQR files,
alternating between Molecules 0 and 1.
Often one wants to tune the reaction criteria to match a known rate constant. Instead of using a trial-and-error approach of setting the required distance in a reaction criterion
(see below ), one can estimate the
reaction rate as a function of required distance in one simulation. This is
done by running a trajectory, and outputting the minimum reaction distance
achieved before escaping for good. The reaction is turned off by setting
the required distance to zero, so that all trajectories escape. Then, for
each value of the distance, the fraction of trajectories that would have
reacted had that distance been the criterion for reaction is computed,
along with the corresponding rate constant. This tabulated function can
be used to find the required distance to give a desired rate constant.
To do this
requires four steps. First, set an output file for the minimum reaction
distances in the body of the input file by including
<min-rxn-dist-file> min-rxn-dist-file-name </min-rxn-dist-file>
Next, set the reaction distances in the reaction description to zero;
this keeps reactions from taking place. Then, run nam_simulation as above. Finally, run the program rates_of_distances, using as
inputs the normal output file of nam_simulation and the file
named inside the min-rxn-dist-file tag:
rates_of_distances -res result-file -dist min-rxn-dist-file-name > output-file
The output gives an XML file with gives the rate constant and its 95%
confidence interval as a function of distance. If you just want to ouput a plain
file for easy plotting, add the flag -plain to the command line.
In order to run a weighted-ensemble simulation, it necessary first to
build configuration space bins; the bin information is placed
in the file prefix0-prefix1-bins.xml . This is done by
running the program build_bins :
build_bins prefix0-prefix1-simulation.xml
The number of system copies used is designated by the
n-bin-copies tag in the input file.
As it runs, reaction coordinate numbers will go scrolling past; they should keepgetting smaller and eventually stop. If that does not happen, i.e.,
the numbers keep on going, you might need to increase the number of system
copies, or it might be that your reaction criterion is unattainable.
After the bins are built, the actual weighted-ensemble simulation is run:
we_simulation prefix0-prefix1-simulation.xml
As before, the results are output to the file specified by
results . In each row of
output numbers, the right-most number is the flux of system copies that
escaped without reaction, while the other ones are reactive fluxes.
So, even for a rare reaction event, you should at least see small numbers for
the reactive fluxes after the system has reacted steady-state.
This can be visually examined at any point, and can also be analyzed as
above, but with a different program:
cat results.xml | compute_rate_constant_we
assuming that results.xml is the results file.
Because the streams of numbers are autocorrelated, a more sophisticated
approach for computing confidence intervals is used (Politis, 1994),
and if there are
not enough data points, the program compute_rate_constant_we will
simply refuse to provide an answer and print out a message about
autocorrelations not dying away quickly enough.
Unlike the single-trajectory method, the weighted-ensemble method
cannot handle arbitrary reaction networks; for now, it can only
handle nested reaction criteria with the same atom-atom contacts
but different required distances.
A reaction criterion is specified by pairs of atoms, with
each atom on
a different molecule. Each pair is also assigned a center-to-center distance.
The reaction itself is assigned a number n.
A pair is said to be "satisfied" if its atoms are closer than
the assigned distance. If n pairs are satisfied,
then the reaction is said to occur. For example, a criterion
might be composed of all pairs of atoms forming hydrogen bonds
in a protein-protein complex, and if any three of the pairs
are within 5.1 Å
In addition to containing
pairs, a reaction criterion can contain other reaction criteria;
these inner criteria are treated like pairs to be satisfied.
An example would be an enzyme with two active sites. The binding
of the substrate to
each site
would be represented by a separate reaction criterion, but the two
reaction criteria could be lumped into another criterion to
represent the overall reaction.
Entire reaction pathways can be specified by named states
and named reactions . As an example,
suppose the system comprises a substrate that can react first at
one active site on an enzyme, and then can react at a second active site.
When the molecules
are initialized for a trajectory, the system is considered to be
in the reactants state, and if they react, the system moves to
the product-1 state.
The reaction itself is calledreaction-1.
The first reaction can then be followed by binding to the other
site. The following state is called product-2, and
the reaction is called reaction-2.
An escape does not count as anything.
The format of the XML file describing the reaction pathway is shown below:
<pathway>
<first-state> reactants </first-state>
<reactions>
....
<reaction>
<name> reaction-1</name>
<state-to> product-1 </state-to>
<state-from> reactants </state-from>
<criterion>
<n-needed> 3 </n-needed>
<pair>
<atoms> 55 111 </atoms>
<distance> 5.1 </distance>
</pair>
....
<criterion> .. </criterion>
....
</criterion>
</reaction>
<reaction>
<number> reaction-2</number>
<state-to> product-2 </state-to>
<state-from> product-1 </state-from>
<criterion>
<n-needed> 3 </n-needed>
<pair>
<atoms> 1109 443 </atoms>
<distance> 5.1 </distance>
</pair>
....
<criterion> .. </criterion>
....
</criterion>
</reaction>
...
</reactions>
</pathway>
Reactions are listed, each with the name, the state before and after,
and the criterion. The criterion lists the number of pairs or
sub-criteria needed, followed by the pairs themselves and any sub-criteria.
Any state that does not have a reaction leading away from it is called
a final state; once a final state is reached, the trajectory is
ended.
Although the above file can be generated by hand, in the case of
two large molecules forming a complex, it is better to have some
help at least to generate the pairs. The program make_rxn_pairs
take four inputs and generates a file listing atom pairs, as shown
in the example:
make_rxn_pairs -mol1 molecule1.pqrxml -mol2 molecule2.pqrxml -ctypes contacts.xml -dist 5.0 > pairs.xml
where the two PQRXML files represent the molecules in their complexed state,
the file contacts.xml denotes the types of atoms involved in
potential contacts. If any two atoms of a possible pair are within
the distance given by the flag -dist , then they are output as
a reaction pair. The output file pairs.xml contains an XML listing
of the reaction pairs. It can be cut-and-pasted into a reaction pathway
file.
If the -nonred flag is present in the input, then after finding possible pairs, the program eliminates redundant pairs. It groups the pairs according to
connectivity, selects the closest bond in each group, and eliminates all
other pairs involving the atoms of the closest pair. It then regroups and
eliminates repeatedly until no more connected groups are left. Previous studies
using SDA used a very similar approach.
The structure of the file contacts.xml can enumerate
pair possibilities in two ways. One way is to list atom types
(which include the residue type) of Molecule 0, and the atom
types of Molecule 1, and state that all combinations of atoms
from the lists is a possible pair. This is done in the
file protein-protein-contacts.xml.bak, where all possible
hydrogen-bonding pairs are implicitly enumerated.
Another way is to explicitly list the pairs. This is the file
structure:
<contacts>
<combinations>
<molecule0>
<contact>
<atom> NE2 </atom>
<residue> HIS </atom>
</contact>
...
</molecule0>
<molecule1>
<contact>
<atom> OD1 </atom>
<residue> ASP </atom>
</contact>
...
</molecule1>
</combinations>
...
<explicit>
<pair>
<contact0>
<atom> ??? </atom>
<residue> ??? </residue>
</contact0>
<contact1>
<atom> ??? </atom>
<residue> ??? </residue>
</contact1>
</pair>
...
</explicit>
</contacts>
The example
file protein-protein-contacts.xml.bak represents
the same rules used by the auxiliary programs in SDA for
protein-protein interactions, and thus can be used for systems
other than thrombin-thrombomodulin.
For the most common case, that of two molecules associating in one
reaction, cutting and pasting pair files into a reaction file
is not necessary. The program
make_rxn_file can be used:
make_rxn_file -pairs pairs.xml -distance 5.0 -nneeded 3 > rxns.xml
This reads in the pairs file pairs.xml and generates a
criterion requiring the atoms of 3 pairs to get within 5 Å of
each other. The criterion is applied to Reaction 1 going
from State 0 to State 1 and output to rxns.xml .
As noted above in the discription of the radii tag, if a sphere has
a radius of zero, it does not physically interact, but can be used to describe
reaction criteria.
These programs are all written in the Ocaml language, with the
exception of born_integral, which is written in C++.
In addition to the
input flags described for each program, if the user uses the input flag
-help, the program will print out a description of itself.
The outputs of these programs eventually funnel into the simulation code
(which is written in C++).
uhbd2dx
Converts UHBD grid file to an OpenDX file. Reads from standard input
and writes to standard output.
pqr2xml
This program converts a PQR file, which
describes the collection of charged spheres making up
the molecule, to an
equivalent XML file (PQRXML file). The reason for this
is that most of the files processed by the UCBD software
are in XML format. PQR files can be generated from PDB
files using software included with APBS. More information on the PQR format, and the equivalent XML format, can
be found in the
APBS documention.
The pqr2xml program receives the PQR file through standard input,
and outputs to standard output. Example:
cat mol.pqr | pqr2xml > mol.xml
xml2pqr
This program does the opposite of pqr2xml , converting a PQRXML file into
a PQR file.
residue_test_charges
This program reads a PQRXML
file from standard input describing the charged spheres (output from
pqr2xml), and outputs a test charge for each residue
that has a total charge significantly greater than zero. Later, I
want to use effective charges, but only after I've implemented
a good way of dealing with their difficulties encountered at close
range.
protein_test_charges
This program reads a PQRXML
file from standard input describing the charged spheres (output from
pqr2xml), and outputs a one or two
test charges for each charged residue, using the method of SDA.
surface_spheres
This program reads a PQRXML
file from standard input describing the charged spheres (output from
pqr2xml),
and outputs an XML file with four lists.
The first list is a list of the triangles of the
surface spheres; each triangle is a trio of integers, with each integer
representing a sphere. These triangles come from an algorithm which
is almost identical to that used in Michel Sanner's MSMS.
A probe rolls across the molecule surface and
touches three spheres at a time as it makes its way.
The second list is a list of integers, each representing a surface sphere.
The third list is a list of "insiders", or those spheres completely
enclosed within a surface sphere.
The fourth list is a list of "danglers", or those spheres that hang out into
the solvent but could not be picked up by the ball-rolling algorithm.
The following input flags are used:
-probe_radius radius of the probe sphere. Default is 1.5.
-direction In order to locate the first surface sphere, the
program must start searching along a particular direction. The default
search direction is (0,0,1); in other words, the program starts with the
sphere whose surface has the greatest z-coordinate. This flag can
change the default direction.
Once the surface spheres and their triangles are computed, the program
must then distinguish between the interior spheres and the danglers.
A point is selected which is guaranteed to outside.
Then, for each remaining sphere, a line segment is constructed
running from the exterior point to the sphere center. The program
counts how many surface triangles are intersected by the line segment
(this is done using a log(n) algorithm so that every triangle does
not need to be checked). The number of intersections denotes whether
the sphere is inside or outside the cage of surface triangles.
If the program is unlucky and the line hits a triangle edge, the program
will perturb the exterior point slightly and try again.
Example:
cat mol.xml | surface_spheres -probe_radius 1.6 -direction 1 0 0 > mol-surface.xml
inside_points
This program outputs an XML file representing a rectangular grid
of points, each with a 1 or 0 depending
on whether the point is inside (1) or outside (0) the molecule.
The program reads in the XML sphere data (from pqr2xml) and
the surface information from surface_spheres.
For this application, a point is "inside" if it meets at one of two criteria.
First of all, if the point's distance from the surface of any surface or
dangler atom is less than a certain exclusion distance set by the user, it is
considered inside. Second, if the point is inside the cage of
triangles formed by the surface spheres, it is
considered to be inside the molecule. The lower corner, spacing, and number
of points in each direction of the grid are set by the user. The 1's and 0's
are output in order, starting from the lower corner, with the x-direction
varying most rapidly. If the lower corner or spacing are not specified,
reasonable and useful defaults are chosen.
The following input flags are used:
-spheres XML file with sphere data
-surface XML file with surface data
(output from surface_spheres)
-corner lower corner of grid
-ngrid number of grid points in each direction
(default 100 100 100)
-spacing spacing between grid points
-exclusion_distance additional interior padding around each
sphere (default 0.0)
Each grid point is tested for inside-ness in same manner as in
surface_spheres above. To speed things up, if a point
has been found to be inside, the distance to the nearest triangle is
found, and all other points within that distance are immediately
marked as "inside" as well. If the point is outside, then the
distance to the nearest sphere surface is found, and the points
within that distance are marked as "outside". This avoids
having to find intersecting triangles for most of the grid points.
The big advantage of this algorithm is that it avoids marking
interior molecular cavities as exterior. This is essential for
efficiently lumping the effective charges together, as seen
below.
Example:
inside_points -spheres mol.xml -surface mol-surface.xml -corner -11.1 22.2 -33.3 -spacing 0.5 -ngrid 65 65 65 -exclusion_distance 1.5 > mol-inside-pts.xml
grid_distances
Given the output of inside_points, this program outputs a
like-shaped grid of distances. If the point from inside_points
is 0, then the distance is that to the nearest point with a value of 1,
and vice versa. The spacing between grid points is obtained from
the output of inside_points, so it is not necessary to
enter it separately. The algorithm time scales as N3/2, where
N is the total number of grid points. The following input flag can
be used:
-plain Instead of an XML file, this reads in a plain
ASCII file.
Example:
cat mol-inside-pts.xml | grid_distances > mol-distances.xml
lumped_charges
The script lumped_charges takes as its input the xml file
of effective charges from compute_effective_charges
and outputs another
xml file containing a hierarchical grouping of the charges.
The key idea is that if the source of electric field
is far away from a group of force centers and you want to compute the
force and torque on the group,
the group can be compressed
into a smaller and faster data structure. Using the technique of
Chebyshev interpolation, the group can be converted into a data
structure that I'll call a chebybox. The chebybox has
a rectangular array of 64 positions (4 by 4 by 4) where the
electric potential
is evaluated. The resulting force is a linear combination of
contributions from each position multiplied by the electric potential evaluated
at the position:
F =
 i=063
Vi fi,
where Vi is the electric potential evaluated at point i
and f i is the contribution from point i.
The torque is computed from a similar linear combination.
Mathematically, the chebybox approximation is exact if the the
potential is a cubic function.
i=063
Vi fi,
where Vi is the electric potential evaluated at point i
and f i is the contribution from point i.
The torque is computed from a similar linear combination.
Mathematically, the chebybox approximation is exact if the the
potential is a cubic function.
Chebyboxes can be nested. If, during the course of a simulation,
the field source comes closer to a group of force centers, then the
chebybox representing the group might not provide accurate forces
and torques, so the group must be split into two groups, each with
its own
chebybox. For example, near a point charge, the potential cannot
be represented by one cubic function over a volume that is large
compared to the distance from the charge. The decision is made
by computing the ratio of distance of the box center
from the field source, to the box diagonal length. If this ratio
is below a certain threshhold, the box is divided.
Finally, it is not worthwhile to use a chebybox if
the number of force centers in a group
is much less than 64; it is better to
evaluate the force on each center explicitly.
In addition to an outer 4 by 4 by 4 chebybox, the program
also generates an outer 3 x 3 x 3 chebybox, which can be used
when the molecules are far apart.
The interior of a large molecule will never get close to another
molecule, so time and space can be saved by not dividing large
chebyboxes buried deep in the molecule. Therefore, one of the
optional inputs is the XML file of distances from grid_distances;
this tells the program how close is the chebybox to the molecule surface.
The following input flags are used:
-pts file of effective charges
-dist optional file of distances
-max maximum number of points in bottom-level box (default 20)
-thr ratio threshhold for dividing boxes (default 2.0)
Example:
lumped_charges -pts mol-charges.xml -dist mol-distances.xml > mol-cheby.xml
compute_effective_volumes
This program computes effective volumes for each sphere in the
molecule. This is needed as an input to the part of the simulation code
that computes the desolvation forces. It estimates the volume of
space closest to each sphere by Monte Carlo integration, but only
includes volume interior to the molecule, as represented by the XML
file output from inside_points. If values for the solvent
and molecule dielectrics are provided, then the output volumes are
scaled in order to function as "effective charges" for computation of
desolvation forces. The following input flags are used:
-spheres file representing spheres (output of pqr2xml)
-inside file representing interior (output of
inside_points)
-solvent solvent dielectric (dimensionless)
-solute solute dielectric (dimensionless)
Example:
compute_effective_volumes -spheres mol.xml -inside mol-inside-pts.xml -solvent 79 -solute 4 > mol-volume.xml
This output file can also be fed into the lumped_charges program to
generate a chebybox structure for computing desolvation forces:
lumped_charges -pts mol-volume.xml -dist mol-distances.xml > mol-vol-cheby.xml
ellipsoids
This program computes two ellipsoids from the effective volume data (output from compute_effective_volumes).
The first ellipsoid is an ellipsoid with the same inertia matrix as the molecule, based on the volume data.
The second ellipsoid is an estimate of the smallest ellipsoid bounding the spheres of the molecule.
This is computed as follows. First, the molecule is stretched and compressed along its principal axes in order
to make it more equivalent to a sphere. Then, the smallest bounding sphere is computed for the sphere centers,
Finally, this bounding sphere is transformed back to make it an ellipsoid, and extra padding is added to account
for the radii of the molecule spheres. These ellipsoids are used in computing hydrodynamic properties
and time step sizes. The output is an XML file.
Example:
cat mol-volumes.xml | ./ellipsoids > mol-ellipsoids.xml
mpole_grid_fit
This program computes a multipole fit (out to quadrupole level) to the outer points of
the input grid. The fit is done by least squares on the surface of the largest
sphere enclosed by the grid. The multipole information is output as an XML file. It also computes the minimum distance from the center at which the field is amost radially symmetric. It can use either an electric field file or a file of charges.
The following
input flags are used:
-dx Electric field in DX format
-xml Electric field in XML format
-charge XML file of molecule charges (output from residue_test_charges or protein_test_charges) if there are no arguments for dx or xml.
-center Molecule center (3 numbers)
-solvdi Solvent dielectric (default 78)
-vperm vacuum permitivity (default value assuming units of Angstroms, electron charge, and kT (298 K))
-debye Debye length
-error do not output fit; only print fractional fitting error
Example:
mpole_grid_fit -dx mol.dx -debye 9.58 > mol-mpole.xml
make_surface_sphere_list
This program generates an XML file (equivalent to PQR) of spheres
on the surface. It reads in the surface and dangler spheres from
the output of surface_spheres, as well as spheres
from the reaction file (output of make_rxn_file) that
have not been included by surface_spheres.
The following input files are used:
-surface surface file (output of surface_spheres
-spheres XML spheres file (output of pqr2xml)
-rxn1 XML reaction file if molecule is first one of pair
-rxn2 XML reaction file if molecule is second one of pair
Example:
make_surface_sphere_list -surface mol1-surface.xml -spheres mol1.xml -rxn1 mol-rxn.xml
make_rxn_file
This program generates an XML file that represents a reaction and its
criteria. The main inputs are the the two PDB files that are outputs from
the da-pairs from the SDA package.
The following input flags are used:
-files two PDB files of reaction atoms
-distance distance required for reaction
-nneeded number of pairs needed for reaction
Example:
make_rxn_file -files mol1.rxna mol2.rxna -distance 2.1 -nneeded 2 > mol-rxn.xml
normalized_rxn_pairs
No longer used.
This program computes the Born integral for a molecule:
 | r - 4 | dr |
| (εp - 1 - εs - 1) | | q2 | | r - 4 | dr |
|
|

32π2ε0 |
-in name of input file, which is the output file of 0's and 1's from inside_points
-vperm vacuum permittivity (default in units of Å, ps, kT at 298 K)
-ieps dielectric of solute (default 4)
-oeps dielectric of solvent (default 78)
-debye Debye length (default infinity)
sphere_return_distribution
This program computes information used by the simulation program to
run the enhanced Northrup-Allison-McCammon algorithm. It contains
the time and angle distribution of trajectories that reach an outer radius
and return to the inner ("b") radius. This enables us to avoid
using a large "q" radius. It is an enhancement of an algorithm of
Luty, Zhou, and McCammon, and will be submitted for publication.
-arad0 radius of Molecule 0
-arad1 radius of Molecule 1
-ch0 charge of Molecule 0
-ch1 charge of Molecule 1
-debye Debye length
-kT temperature times Boltzmann constant (default 1)
-vperm vacuum permittivity (default in units of Å, ps, kT at 298 K)
-diel solvent dielectric (default 78)
-h2ovisc water viscosity at 298 K (default in units of Å, ps, kT at 298 K)
-visc viscosity relative to water at 298 K (default 1)
-rbegin beginning radius of trajectory. Ending radius is 1.1 times the beginning radius
compute_charges_squared
This program computes the square of the effective charges. It reads in a charge file
from standard input, and outputs to standard output in the same XML format.
This is used in computing the desolvation forces.
hydro_radius
This program computes the hydrodynamic radius of the molecule by computing
the mean chord length (Hansen, 2004)
make_soft_spheres
This program receives a pqrxml file through standard input, and
outputs the same information with some of the spheres marked as
being "soft", or subject to soft short-ranged interactions.
-soft Another pqrxml file containing only the spheres
to be marked as soft. It compares the number tag of
the spheres in the two files.
compute_rate_constant
This program takes as input the results file from nam_simulation and outputs the rates and their confidence intervals.
compute_rate_constant_we
This program takes as input the results file from we_simulation and outputs the rates and their confidence intervals.
make_repul_params
This takes a molecule pqrxml file from standard input, and outputs a short-ranged force parameter file with parameters that
give only the repulsive r-12 part of the Lennard-Jones force. The value of ε provided causes the interaction
energy to be equal to kT (with T = 298 K) when the sphere centers are separated by the sum of their radii. (Because of the nature
of the parameterization, this is done approximately, but accurately,
by using small values of ε and large values of the radius parameter).
process_trajectories
- see above.
xyz_trajectory
- see above.
These are files generated for the Molecule 0. In the file names, prefix0 is replaced
by the actual prefix given in the input file to bd_top.
- prefix0-charges.xml - a set of effective charges
- prefix0-cheby.xml - Chebyshev interpolation data structure for
effective charges
- prefix0-distances.dx - a dx file containing the distances of
each exterior point to the nearest interior point, and vice-versa
- prefix0-ellipsoids.xml - bounding ellipoid using for estimating
hydrodynamic interactions
- prefix0-hydro-radius.xml - hydrodynamic radius
- prefix0-q2.xml - effective charges squared; used for the desolvation forces
- prefix0-q2-cheby.xml - Chebyshev interpolation data structure for
effective charges squared
- prefix0-surface.xml - a list of atoms on the surface found by
rolling a probe sphere.
- prefix0-surface-atoms.xml - actual list of surface atoms to be used
for collision detection
- prefix0-volume.xml - list of effective volumes of each atom
- prefix0-mpole.xml - multipole expansion of effective charges
For Molecule 0, there are one or more OpenDX electric potential
files from APBS. For each,
there is generated a file ins-potential.xml ,
where potential.dx is the file from APBS.
This is a grid file of integers
(but in XML format) which denotes the interior (value of 1)
and
exterior points (value of 0) of the molecule.
Likewise, for each file from APBS is generated
a file born-potential.dx ; this is a grid of Born integral
values used for the desolvation forces.
An analogous set of the above files is generated for Molecule 1.
The following files are generated for both molecules:
- prefix0-prefix1-solvent.xml - solvent information
- prefix0-prefix1-old-data.xml - previous data, used for detecting changes in parameters
- prefix0-prefix1-normed-rxns.xml - because only the surface atoms are read into the simulation programs, the atoms in the reaction criteria must be renumbered so that they are in sequence. This is the resulting file.
- prefix0-prefix1-return-dist.xml - information used for appropriately returning the molecule to the b-sphere; has data on the time and probability distributions.
- prefix0-prefix1-simulation.xml - input file into the simulation programs.
A tutorial with an example is available
here .
To-Do List
- finish hollowed-out grid option for saving memory
- effective Born forces for close range with smooth segue from more accurate effective charge model
- Flexible bodies with internal-coordinate BD
This work is copyrighted under the terms of the
MIT License .
The actual notice is here .
I am grateful to Andy McCammon for providing the necessary
infrastructure and guidance for this project, to
Adam Van Wynsberghe for helpful discussions and
providing data for the thrombin-thrombomodulin example, and to
Maciej Dlugosz for patiently enduring a first draft of this software
and providing invaluable feedback. Barry Grant also has endured
various incarnations of the software and provided very helpful
suggestions. I am also grateful to the
authors of
Expat,
Ocaml-Expat,
CLAPACK,
and the random number generator.
This work is supported by the Howard Hughes
Medical Foundation.
Ermak, DL and McCammon, JA. Brownian Dynamics with Hydrodynamic Interactions,
J. Chem. Phys. 69, 1352-1360 (1978)
Northrup SH, Allison SA and McCammon JA.
Brownian Dynamics Simulation of Diffusion-Influenced Bimolecular Reactions
J. Chem. Phys. 80, 1517-1526
Luty BA, McCammon JA and Zhou HX. Diffusive Reaction-Rates From Brownian Dynamics Simulations - Replacing the Outer Cutoff Surface by an Analytical Treatment,
J. Chem. Phys. 97, 5682-5686 (1992)
Huber GA and Kim S. Weighted-ensemble Brownian dynamics simulations for protein association reactions, Biophys. J 70, 97-110 (1996)
Gabdoulline RR and Wade RC. Effective charges for macromolecules in solvent,
J. Phys. Chem 100, 3868-3878 (1996)
Hansen S.
Translational friction coefficients for cylinders of arbitrary axial ratios estimated by Monte Carlo simulation,
J. Chem. Phys. 121, 9111-9115 (2004)
Politis DN and Romano JP. "The Stationary Bootstrap", J. Amer. Statist. Assoc. 89, 1303-1313 (1994)